Grant Applications/CME content
New and Emerging Therapies to Manage Type 2 Diabetes Mellitus
Learning Objectives
Upon completion of this program participants will be able to:
-
Describe the unmet needs associated with traditional treatments of type 2 diabetes mellitus (T2DM)
-
Describe the physiological effects of incretins on glycemic control
-
List the incretin based treatments of T2DM and describe their mechanisms of action efficacy and adverse effects
-
Integrate incretin based treatments of T2DM into their practice
Needs Assessment
Challenges in the Treatment of Type 2 Diabetes Mellitus
Type 2 diabetes mellitus has a high and increasing prevalence in the US. Based on the 2005 National Health and Nutrition Examination Surveys (NHANES), an estimated 20 million people in the United States had diabetes. Of these 90-95% had Type 2 diabetes mellitus and approximately 30% had undiagnosed cases 1,2. The number of diagnosed cases is expected to triple by 20503. In 2002 diabetes was the 6th leading cause of death1,4 and associated with $92 billion in direct and $40 billion in indirect costs1.
The development of complications in diabetes is strongly associated with the glycemic level. Effective glycemic control can reduce a patient's risk for microvascular complications including retinopathy, nephropathy, and neuropathy. However, patients with higher glycosylated hemoglobin (Hb A1c) levels at diagnosis as well as those experiencing and upward drift of Hb A1c levels over time were at an increased risk of complications as evidenced by higher healthcare costs.5
The goal in diabetes treatment is to maintain Hb A1c levels below 7%.6 However, this goal is not routinely achieved in medical practice,7,8 and several barriers to normalizing glycemia exist. First, established diabetes treatments poorly control postprandial hyperglycemia, limiting the effort to restore Hb A1c levels to the non-diabetic 4-6% range.9 Second, treatment with insulin or oral secretagogue may lead to hyperglycemia, thereby putting patients at risk for accidental injuries and limiting the willingness of both physicians and patients to intensify treatment.10,11 Third, treatment with insulin, secretagogues, and thiazolidinediones may cause weight gain of up to 8 Kg with intensive insulin treatment of T2DM.10,12 Finally, oral agents are subject to loss of efficacy over time.10
Pathophysiological Mechanisms and Novel Drug Targets
Traditionally diabetes has been described in terms of insulin secretion and hepatic sensitivity to insulin.10 While this description is most accurate in the fasted state, avoiding postprandial hyperglycemia requires a rapid and sustained release of insulin.13 This enhanced release of insulin is mediated by gut-related factors termed incretins, in particular amylin and the glucagon-like peptide 1 (GLP-1).10,14,15
Amylin is a 37–amino acid peptide that is co-secreted together with insulin from islet β-cells. Its plasma levels increase in response to food intake.10Exogenous amylin inhibits both gastric emptying and glucagon secretion, which in turn leads to reduced food intake and weight loss.16 GLP-1 is a 30 –amino acid gut peptide produced in enteroendocrine L cells located in the distal ileum and colon. GLP-1 is rapidly secreted from the distal gut within minutes of food ingestion. The effects of GLP-1 secretion include stimulation of insulin secretion, inhibition of both gastric emptying and glucagon secretion as as well as activation of CNS regions involved in the control of satiety..14,17 In preclinical models of experimental diabetes, GLP-1 also induces an expansion of β-cell mass via stimulation of cell proliferation and inhibition of apoptosis.18,19
In keeping with these physiological properties, administration of native GLP-1(7–36) amide or GLP-1(7–37) inhibits gastric emptying and glucagon secretion and potentiates of glucose-dependent insulin secretion and thereby limits postprandial hyperglycemia.14,15Exogenous GLP-1 also reduces food intake and enhances satiety in normal and diabetic subjects20 and induces weight loss in healthy obese subjects.21Native GLP-1 is substrate to cleavage by dipeptidyl peptidase 4(DPP-4) which together with renal clearance limits its half life to several minutes.22Because of this short half life, pharmacological strategies for enhancing GLP-1 action involve either the development of GLP-1R agonists that are not subject to enzymatic cleavage or increasing the levels of circulation native GLP-1 by inhibition of DPP-4.10
Efficacy, Safety and Clinical Application of Incretin Based Treatments for Type 2 Diabetes Mellitus
Pharmacological strategies that enhance the action of incretins have to avoid the biological limitations of the native molecules. In the case of amylin a synthetic analogue had to be developed to avoid the potential of fibril formation found in human amylin. The synthetic analogue, pramlitide has three amino acid substitutions that prevent fibril formation but do not impair the the biological potency of the molecule.10Pramlitide lowers glucose excursions after meals and also reduces appetite; it is approved for the treatment of type 1 and insulin-requiring type 2 diabetes.10,23
Two different drug classes have been developed to potentiate the effects of GLP-1, namely degradation resistant GLP-1R agonists and DDP-4 in inhibitors which suppress the enzymatic inactivation of GLP-1. GLP-1R agonists are delivered via subcutaneous injection whereas DDP-4 inhibitors can be taken orally.24
Exenatide is the first The first GLP-1R agonist to be approved for human clinical use.24 It mimics the full spectrum of GLP-1R dependent actions including slowing of gastric emptying, suppression of glucagon secretion, promotion of satiety and nutrient-stimulated insulin secretion, but is resistant to cleavage by DDP-4. The most prominent side effects are nausea and vomiting, especially at the beginning of treatment. 10,24In a head to head comparison with insulin, exanatide achieved comparable glycemic control, but whereas patients on insulin showed a mean weight gain of 1.8 Kg, a mean weight loss of 2.3 Kg was observed in patients on exenatide.25To date, the evidence form clinical trials suggests that exenatide represents a reasonable alternative to insulin therapy in patients whose diabetic symptoms are suboptimally controlled with oral hypoglycemic agents, particularly for those who are concerned about weight gain. However, the available data do not yet support the hypothesis that therapy with GLP-1R agonists produces durable improvements in β-cell function.24
Liraglutide was developed to have phamacokinetic properties that allow once daily injection by binding noncovalently to albumin.10,24 The efficacy of liraglutide versus that of exenatide, or insulin was investigated in several phase III clinical trials. The results suggest that liraglutide was at least as efficacious in lowering HbA1c as comparator treatments and was usually associated with weight loss of several kilograms. As with exenatide, nausea is the most common adverse effect associated with liraglutide administration.24
In addition to nausea and vomiting, the formation of antibodies and pancreatitis are known side effects of GLP-1R agonists.24While the presence of antibodies does not appear to be a major determinant of effectiveness, pancreatitis is of more concern. Pancreatitis has been reported form post-marketing surveillance of exenatide and during the liraglutide clinical test program. As of 2009, at least 6 deaths have occurred in GLP-1R agonist treated patients due to pancreatitis.24,26
DPP-4 inhibitors suppress enzymatic cleavage of GLP-1 and so exert their glucose-regulatory actions through prolongation of the actions of native GLP-1. The DPP-4 inhibitor sitagliptin was approved in the US in 2006 for use as monotherapy or in combination with metformin, or a sulfonylurea, or a thiazolidinedione.27-31In clinical trials, a significant proportion of patients treated with sitagliptin achieved their target Hb A1c levels. Sitagliptin can be taken orally once daily, and it does not influence body weight.24
The increased number of choices for treating type 2 diabetes mellitus has caused heightened uncertainty to physicians and patients regarding the most appropriate way to treat the disease.32,33
The Consensus Algorithm for the Initiation and Adjustment of Therapy by the American Diabetes Association and the European Association for the Study of Diabetes33 divides the available therapies for diabetes into a well validated tier 1, a less well validated tier 2 therapies and other therapies. GLP-1R agonists are ranked as tier 2 therapies because of their unknown long tern safety profile. They are however included in the treatment algorithm for T2DM, as an adjunctive therapy to be used when diabetes is not well managed by lifestyle and metformin. The choice of a GLP-1R agonist is appropriate when hypoglycemia is particularly undesirable e.g. in patients with hazardous jobs.
Unlike GLP-1R agonists, DPP-4 inhibitors are not listed in the 2 tiers of preferred treatments, because of their lower or equivalent overall glucose-lowering effectiveness compared with the first- and second-tier agents, limited clinical data and relative expense.33 Nevertheless, DPP-4 inhibitors may be appropriate for selected patients.
Program Outline
Talks
1. Current Challenges in the Management of Type 2 Diabetes Mellitus
-
Give an overview of epidemiological data and socioeconomic cost
-
State treatment goals for T2DM and show where traditional treatments fall short
-
control of postprandial hyperglycemia
-
avoiding hypoglycemia
-
loss of effectiveness
-
weight gain
-
2. The Pathophysiology of Type 2 Diabetes Mellitus Reexamined
-
Briefly describe T2DM pathophysiology in the fasted state
-
Describe the physiological functions of incretins
-
Describe the effects of administration and blockade of native incretins
3. An Introduction to Incretin Based Treatments of Type 2 Diabetes Mellitus
-
Introduce the various classes of incretin based drugs on the market and in late stage development
-
discuss mechanisms of action
-
Show efficacy and safety data from clinical trials
-
Explain how incretin based drugs fit into the treatment algorithm for T2DM and the considerations for their use
Enduring Activity
New and Emerging Therapies to Manage Type 2 Diabetes Mellitus
-
Record a moderated round table discussion with the presenters of the talks proposed above. The discussion will touch on
-
The shortcomings of traditional T2DM treatments
-
The physiological functions of incretins
-
Incretin based treatments of T2DM
-
Treatment algorithms for T2DM that include incretin based options
-
1. Deshpande AD, Harris-Hayes M, Schootman M. Epidemiology of Diabetes and Diabetes-Related Complications. Physical Therapy. 2008;88(11):1254 -1264.
2. NHANES - National Health and Nutrition Examination Survey Homepage. Available at: http://www.cdc.gov/nchs/nhanes.htm. Accessed July 24, 2011.
3. Narayan KMV. Impact of Recent Increase in Incidence on Future Diabetes Burden: U.S., 2005-2050. Diabetes Care. 2006;29(9):2114-2116.
4. Centers for Disease Control. ndfs_2005.pdf. Available at: http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2005.pdf. Accessed July 24, 2011.
5. Caro JJ, Ward AJ, O’Brien JA. Lifetime costs of complications resulting from type 2 diabetes in the U.S. Diabetes Care. 2002;25(3):476-481.
6. American Diabetes Association. Standards of Medical Care in Diabetes. Diabetes Care. 2005;28(suppl_1):S4-S36.
7. Gillies CL, Abrams KR, Lambert PC, et al. Pharmacological and lifestyle interventions to prevent or delay type 2 diabetes in people with impaired glucose tolerance: systematic review and meta-analysis. BMJ. 2007;334(7588):299-299.
8. Brown JB, Nichols GA, Perry A. The Burden of Treatment Failure in Type 2 Diabetes. Diabetes Care. 2004;27(7):1535 -1540.
9. Monnier L, Lapinski H, Colette C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycemia of type 2 diabetic patients: variations with increasing levels of HbA(1c). Diabetes Care. 2003;26(3):881-885.
10. Riddle MC, Drucker DJ. Emerging Therapies Mimicking the Effects of Amylin and Glucagon-Like Peptide 1. Diabetes Care. 2006;29(2):435 -449.
11. Cryer PE. Hypoglycemia is the limiting factor in the management of diabetes. Diabetes Metab. Res. Rev. 1999;15(1):42-46.
12. Lindström T, Eriksson P, Olsson AG, Arnqvist HJ. Long-term improvement of glycemic control by insulin treatment in NIDDM patients with secondary failure. Diabetes Care. 1994;17(7):719-721.
13. Caumo A, Luzi L. First-phase insulin secretion: does it exist in real life? Considerations on shape and function. American Journal of Physiology - Endocrinology And Metabolism. 2004;287(3):E371 -E385.
14. Drucker DJ. Enhancing Incretin Action for the Treatment of Type 2 Diabetes. Diabetes Care. 2003;26(10):2929 -2940.
15. Deacon CF. Therapeutic Strategies Based on Glucagon-Like Peptide 1. Diabetes. 2004;53(9):2181 -2189.
16. Schmitz O, Brock B, Rungby J. Amylin Agonists: A Novel Approach in the Treatment of Diabetes. Diabetes. 2004;53(suppl_3):S233-S238.
17. Turton MD, O’Shea D, Gunn I, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. 1996;379(6560):69-72.
18. Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48(12):2270 -2276.
19. Li Y, Hansotia T, Yusta B, et al. Glucagon-like Peptide-1 Receptor Signaling Modulates β Cell Apoptosis. Journal of Biological Chemistry. 2003;278(1):471 -478.
20. Verdich C, Flint A, Gutzwiller J-P, et al. A Meta-Analysis of the Effect of Glucagon-Like Peptide-1 (7–36) Amide on Ad Libitum Energy Intake in Humans. Journal of Clinical Endocrinology & Metabolism. 2001;86(9):4382 -4389.
21. Näslund E, King N, Mansten S, et al. Prandial subcutaneous injections of glucagon-like peptide-1 cause weight loss in obese human subjects. Br. J. Nutr. 2004;91(3):439-446.
22. Orskov C, Wettergren A, Holst JJ. Biological effects and metabolic rates of glucagonlike peptide-1 7-36 amide and glucagonlike peptide-1 7-37 in healthy subjects are indistinguishable. Diabetes. 1993;42(5):658-661.
23. Chapman I, Parker B, Doran S, et al. Effect of pramlintide on satiety and food intake in obese subjects and subjects with type 2 diabetes. Diabetologia. 2005;48(5):838-848.
24. Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5(5):262-269.
25. Heine RJ, Van Gaal LF, Johns D, et al. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann. Intern. Med. 2005;143(8):559-569.
26. Ahmad SR, Swann J. Exenatide and rare adverse events. N. Engl. J. Med. 2008;358(18):1970-1971; discussion 1971-1972.
27. Aschner P, Kipnes MS, Lunceford JK, et al. Effect of the Dipeptidyl Peptidase-4 Inhibitor Sitagliptin as Monotherapy on Glycemic Control in Patients With Type 2 Diabetes. Diabetes Care. 2006;29(12):2632 -2637.
28. Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and Safety of the Dipeptidyl Peptidase-4 Inhibitor Sitagliptin Added to Ongoing Metformin Therapy in Patients With Type 2 Diabetes Inadequately Controlled With Metformin Alone. Diabetes Care. 2006;29(12):2638 -2643.
29. Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE. Effect of Initial Combination Therapy With Sitagliptin, a Dipeptidyl Peptidase-4 Inhibitor, and Metformin on Glycemic Control in Patients With Type 2 Diabetes. Diabetes Care. 2007;30(8):1979 -1987.
30. Scott R, Loeys T, Davies MJ, Engel SS. Efficacy and safety of sitagliptin when added to ongoing metformin therapy in patients with type 2 diabetes. Diabetes Obes Metab. 2008;10(10):959-969.
31. Raz I, Chen Y, Wu M, et al. Efficacy and safety of sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes. Curr Med Res Opin. 2008;24(2):537-550.
32. Nathan DM. Finding new treatments for diabetes--how many, how fast... how good? N. Engl. J. Med. 2007;356(5):437-440.
33. Nathan DM, Buse JB, Davidson MB, et al. Medical Management of Hyperglycemia in Type 2 Diabetes: A Consensus Algorithm for the Initiation and Adjustment of Therapy. Clinical Diabetes. 2009;27(1):4 -16.
Writing for Peer Review
The Clinical Efficacy and Safety of Bortezomib in the Treatment of Relapsed Multiple Myeloma
Multiple myeloma is a hematologic malignancy of B-cell origin and is generally considered incurable.1 There is currently no standard treatment for relapsed multiple myeloma, but the use of high dose dexamethasone is common.2
The proteasome inhibitor bortezomib (VELCADE; Ortho Biotech, a division of Janssen-Cilag, Beerse, Belgium; and Millennium Pharmaceuticals, Cambridge, Massachusetts, USA) is indicated as a monotherapy for the treatment of progressive multiple myeloma in patients who have received at least one prior therapy and who have already undergone or are unsuitable for bone marrow transplantation.1
Two phase II trials demonstrated the efficacy of bortezomib as a treatment for relapsed multiple myeloma and established the dose regimen as 1.3 mg/m2 on days 1, 4, 8, and 11 in 3-week cycles.1 Approval of bortezomib was based on the multinational, randomized phase III Assessment of Proteasome Inhibition for Extending Remissions (APEX) trial.1,3 The APEX trial compared a regimen of 1.3 mg/m2 bortezomib given as an intravenous bolus to high dose dexamethasone (40 mg orally).2 The trial showed that patients treated with bortezomib had longer time to progression as well as significantly higher response and one year survival rates than patients treated with dexamethasone. For detailed results see Table 1.
The safety population of the APEX trial comprised 663 patients, 331 on bortezomib and 332 on dexamethasone. The duration of treatment did not differ significantly between the bortezomib and dexamethasone groups. Early discontinuation for adverse events occurred in 121 patients (37%) in the bortezomib group.2 Adverse events leading to discontinuation included peripheral neuropathy (8%) and thrombocytopenia, various gastrointestinal disorders, fatigue, hypercalcemia, and spinal cord compression (2 % each). In the dexamethasone group, discontinuation for adverse events occurred in 96 patients (29%), and adverse events leading to discontinuation included psychotic disorder, hyperglycemia, or thrombocytopenia (2 % each).
An extended analysis of the APEX trial data with a mean follow up of 22 months showed an overall survival of 29.8 months (95% CI: 23.2–not evaluable) in the bortezomib arm versus 23.7 months (95% CI: 18.7-29.1) in the dexamethasone arm (hazard ratio .77, P .027).4 In the extended analysis, overall and complete response rates were 43% and 9%, respectively. Among responders, 56% showed an improvement of response quality with longer therapy. The extended analysis did not identify any new safety concerns, nor did it yield any evidence of new cumulative toxicity.
An exploratory cohort analysis of the bortezomib arm of the APEX study revealed that patients achieving complete response had a significantly longer median treatment free interval and longer time to alternative therapy than patients achieving very good partial response, see Figure 1.
Bortezomib is the only single agent to have shown a survival benefit in patients with relapsed multiple myeloma.3 The sum of data form phase II and phase III trials as well as an extension study indicate indicate that prolonged therapy is associated with a manageable safety profile.4
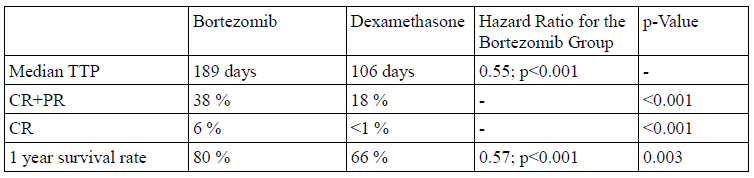
Table 1: Results of the APEX trial.2 Abbreviations: TTP, time to progression; CR, complete response; PR, partial response
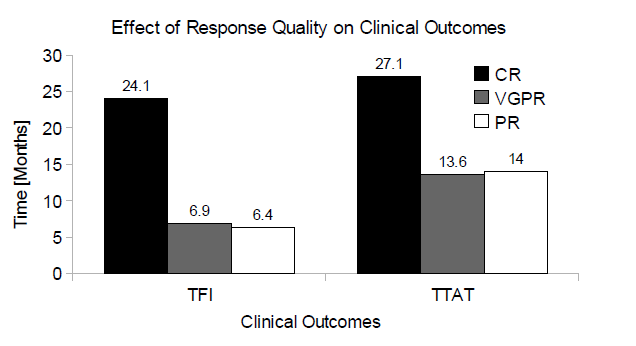
Figure 1: The effect of response quality on clinical outcomes. Abbreviations: CR, complete response; VGPR, very good partial response; PR, partial response; TFI, treatment free interval; TTAT, time to alternative treatment
1. Armand J, Burnett AK, Drach J, et al. The emerging role of targeted therapy for hematologic malignancies: update on bortezomib and tipifarnib. Oncologist. 2007;12(3):281-290.
2. Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005;352(24):2487-2498.
3. Niesvizky R, Richardson PG, Rajkumar SV, et al. The relationship between quality of response and clinical benefit for patients treated on the bortezomib arm of the international, randomized, phase 3 APEX trial in relapsed multiple myeloma. Br. J. Haematol. 2008;143(1):46-53.
4. Richardson PG, Sonneveld P, Schuster M, et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final time-to-event results of the APEX trial. Blood. 2007;110(10):3557-3560.
News Writing
First Test to Distinguish MRSA from MSSA
The KeyPath MRSA/MSSA Blood Culture Test by MicroPhage Inc. was cleared by the FDA
Alex Kadner, PhD
May 9, 2011 – According to a news release, the FDA has cleared the first test to distinguish methicillin resistant Staphylococcus aureus (MRSA) from methicillin susceptible Staphylococcus aureus (MSSA). The test is manufactured by MicroPhage Inc. of Longmont, CO and marketed as the KeyPath MRSA/MSSA Blood Culture Test. The FDA decision to clear the test was based on 1116 samples that were tested at four major US treatment centers.
The test accurately identified MRSA in 98.9% (178/180) of cases, and MSSA in 99.4% (153/154) of cases. The test takes about 5 hours to complete after bacterial growth in the blood sample is first detected and does not require specific instruments other than blood culture equipment.
Staphylococci bacteria cause many types of infection including skin, blood and food infections as well as pneumonia. These infections are usually treated with antibiotics. However, MRSA infections are resistant to commonly prescribed antibiotics such as penicillin and amoxicillin. MRSA infections may occur anywhere, but are particularly dangerous and possibly life threatening in healthcare settings, because patients there may have weakened immune systems and may undergo procedures such as surgery that allow the spread of MRSA from the skin into the body.
According to the Committee to Reduce Infection Deaths (www.hospitalinfection.org), MRSA can live for many hours on surfaces and fabrics. Since MRSA does not cause any illness unless introduced into the body, for example via an open wound or a catheter, carriers can unknowingly introduce MRSA into healthcare settings. To stop the spread of MRSA in healthcare settings, these carriers must be identified.
Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics Device Evaluation and Safety in the FDA's Center for Devices and Radiological Health, stated that "Clearing this test gives health care professionals a test that can confirm S. aureus and then identify whether the bacteria is MRSA or MSSA," He added “This not only saves time in diagnosing potentially life-threatening infections but also allows health care professionals to optimize treatment and start appropriate contact precautions to prevent the spread of the organism."
Additional samples are available by request.